 hotline服务热线:010-61006450
hotline服务热线:010-61006450
解析 | 儿童用药相关政策法规汇总分析-凯发k8旗舰厅
据2020年世界各国儿童(14岁及以下)人口总数统计,全世界儿童人口约为19.76亿,我国儿童人口比例占世界约12.6%,儿童人口基数大,与之不匹配的儿童用药稀缺,儿童用药市场亟需拓展,为此国家出台系列相关法规,便于企业及机构能够加快儿童用药研发进展,为广大儿童的用药健康提供保障。
背景概述
根据人用药物注册技术要求国际协调会(ich)定义:
-
早产新生儿
-
足月新生儿——0-27天
-
婴幼儿——28天-23个月
-
儿童——24个月-11周岁
-
青少年——12-17周岁
市场现状
-
3500多种药品中,供儿童专用的不足60种,1.7%;
-
18万条药品注册批件中,儿童专用药3000多条,涉及400个品种,占比不足2%;且主要以颗粒剂为主;
-
全国6000多家药品生产企业,其中涉及儿童药生产企业1000多家,专门生产儿童用药企业不足10家,占比不足1%。
-
抗菌药物不合理使用
-
静脉注射过渡使用
-
中药安全性问题
-
超说明书用药
《关于改革药品医疗器械审评审批制度的意见》国发〔2015〕44号
加快转移到境内生产的创新药和儿童药的审评审批。
《临床急需儿童用药申请优先审评审批的品种评定的基本原则 》
新增用于儿童人群品种:
-
针对严重威胁儿童生命或者影响儿童生长发育,且目前无有效治疗药物或治疗手段的疾病;
-
相比现有的治疗药物,具有明显治疗优势。
改剂型或新增规格品种:
-
国内现行药品说明书中包含有确定的“儿童用法用量”;
-
现有剂型或规格均不适用于儿童,新增剂型或规格适合于儿童。
仿制品种:
-
对于目前市场短缺的儿童用药品。
2017.12.27
《关于鼓励药品创新试行优先审评审批的意见》 食药监药化管(2017)126号
防治下列疾病且具有明显临床优势的药品注册申请:艾滋病;肺结核;病毒性肝炎;罕见病;恶性肿瘤;儿童用药品;老年人特有和多发的疾病。
《中华人民共和国药品管理法》
鼓励儿童药品研制和创新,支持开发符合儿童生理特征的儿童用药新品种、剂型和规格。
《药品注册管理办法》
第六十八条 药品上市许可申请时,以下具有明显临床价值的药品,可以申请适用优先审评审批程序:(二)符合儿童生理特征的儿童用药品新品种、剂型和规格。
《药品上市许可优先审评审批工作程序(试行) 》
适用范围:
-
临床急需的短缺药品、防治重大传染病和罕见病等疾病的创新药和改良型新药;
-
符合儿童生理特征的儿童用药品新品种、剂型和规格;
-
疾病预防、控制急需的疫苗和创新疫苗;
-
纳入突破性治疗药物程序的药品;
-
符合附条件批准的药品;
-
国家药品监督管理局规定其他优先审评审批的情形。
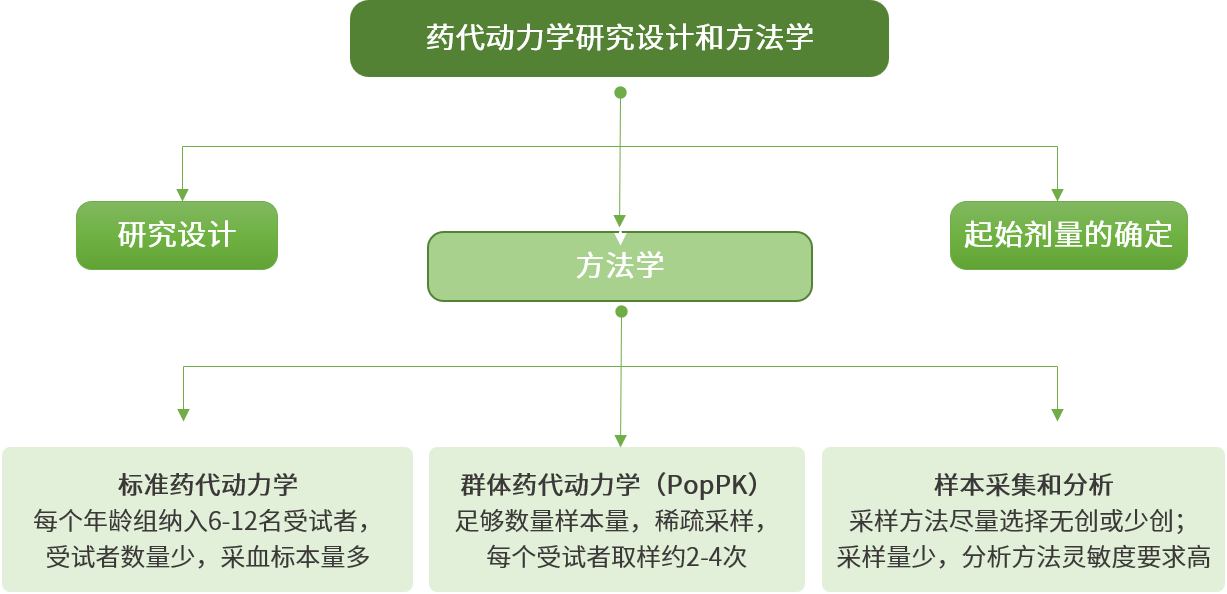
知情同意:
-
低年龄段的儿科人群,须征得其法定监护人的知情同意并签署知情同意书。
-
当儿童有知情同意的能力可以作出参加研究的决定时,还必须同时征得其本人同意。
风险最小化原则:
-
要求研究人员接受良好培训且有丰富的儿科研究经验,包括具备评估和处理潜在的儿科人群不良事件的能力。
-
应事先建立应急机制确保在发现未预期的风险时能够迅速中止研究和实施补救措施。
痛苦最小化原则:
-
研究实施中应充分考虑到儿科受试者的心理感受,加强安抚,营造合适的环境,最大程度减少儿科受试者的痛苦。
2、《儿科人群药物临床试验技术指导原则》
伦理委员会:儿科药学;儿科临床医学;儿童心理学;律师;社区代表(老师、育有子女父母)。
试验机构:cfda认证的儿科人群药物临床试验机构。
情形一:拟用于儿科特有疾病或患者主要为儿科人群的疾病的药物。
-
如果成人无法提供充分信息,则在获得了成人的初步安全性和药代动力学数据之后,即可在目标年龄段儿科人群中开展临床试验。
情形二:拟用于成人和儿科人群共患疾病的药物。
-
如果该疾病是目前缺乏有效治疗的危重症或进展性预后不良疾病,应考虑在获得成人初步安全性及潜在获益的临床试验数据后,例如ii期结束或完成概念验证性研究后,尽早地开展儿科人群临床试验;
-
如果该疾病已有可选择的治疗药物,应在成人iii期确证性研究证明了其在成人患者中的获益大于风险后,再启动儿科人群临床试验;
-
如果预期有较大的安全性风险,建议在该药品成人应用上市后获得充分的安全性数据时再开展儿科人群药物临床试验。
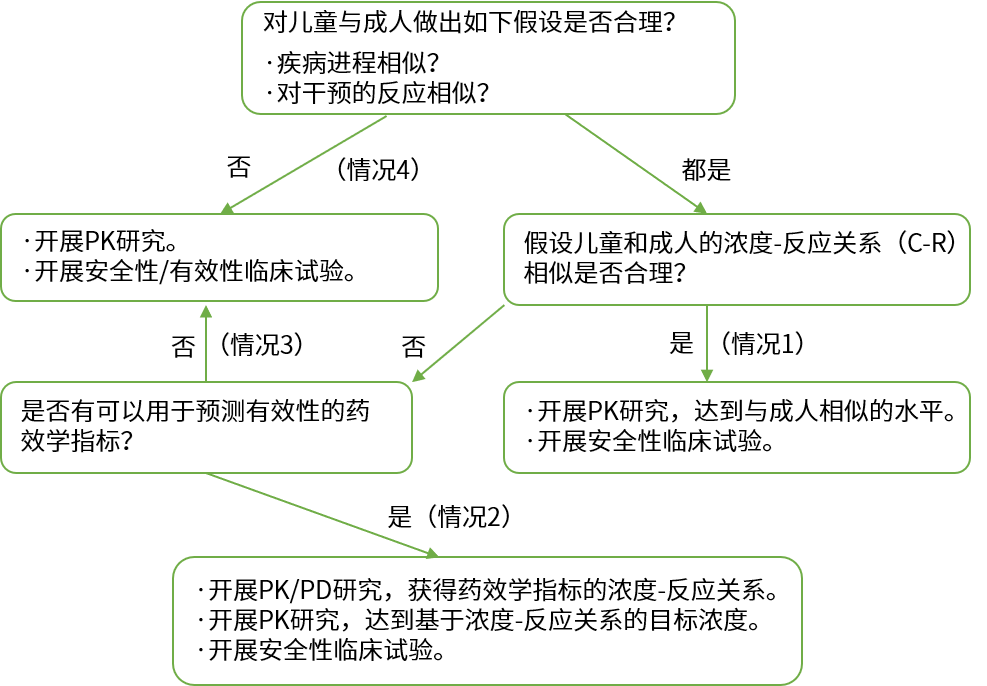
原则:成人临床试验数据向儿科人群的外推限于疗效数据;儿科人群安全性数据需要在儿科人群中开展试验。
决策:目标适应症的疾病进程和治疗反应在成人和儿科人群间是否相似;.药物的体内暴露效应关系(exposure-response relationship)在成人和儿科人群间是否相似。
相似:pk确认或通过外推获得剂量,在特定的儿科人群开展随机对照试验,重点是获得该人群的安全性数据,同时,可以验证拟定剂量的合理性。
不相似:不具备外推成人疗效数据的条件,需开展全面系统的儿科人群药物临床试验。
评价指标的选择:需要良好配合和充分理解的指标,例如,疼痛评估、肺功能检查。
安慰剂对照的设立:
-
当没有其他可接受的治疗方法,受试药物是首个可能有效的药物时;
-
当常规使用的治疗方法的有效性未得到确证时;
-
当常规使用的治疗方法伴随严重的、高发的不良反应,且风险可能明显大于获益时;
-
当用于证明一种已被确证疗效的治疗附加另一种新的治疗后的有效性时;
-
疾病的进程具有不确定性时,例如自发恶化或缓解;
-
需要确定药物的绝对有效性时。
生长发育的监测:儿科人群药物临床试验的随访时间通常较成人试验长,用以观察对生长发育的影响。
主旨:数据外推主要针对有效性数据,以获得明确的剂量。
适用范围:已有中国成人数据的产品外推至中国儿科人群,无中国成人数据产品的外推不在本指导原则中体现。
概念:通过科学的研究方法,将已知中国成人的研究信息和结论,扩展到未知的儿科人群(目标人群)从而减少在未知儿科人群开展不必要的研究。
建立外推假设:整合已知数据,评价已知人群与目标人群的相似性和差异点,借助建模模拟的方法,明确提出外推假设,获得预测指标。
设计外推计划:基于外推假设,制定目标人群研究计划,包括哪些数据可直接通过外推获得,哪些需设计简化的临床试验或完整系统的临床试验获得。
实施外推分析:解释在目标人群中获得的有限数据,验证外推假设,确证/验证已知人群和目标人群相似性。
附:美国食品药品管理局(fda)的儿科人群研究设计与外推决策流程图
4、《儿童用药(化学药品)药学开发指导原则(试行)》
信息收集与分析:首先收集关于该药物的已有研究信息,例如成人的药代动力学和部分药效学数据、非临床研究数据、原料药的关键理化特性、原料药和成人用制剂的质量属性和稳定性等。
初步选择合适的给药途径和剂型:鼓励申请人基于儿童人群特点和临床需求,开发适宜儿童使用的新剂型。
拟定剂型,确定目标产品质量情况:应对药物安全性、有效性和患者可接受性等方面进行重点评。
产品开发实施:建议特别关注儿童患者的可接受性,包括包装系统、给药装置和量取装置的适用性和合理性等。
附药物临床试验相关英文缩写名词解释:
ba:bioavailability,生物利用度
be:bioequivalence,生物等效性
bsa:body surface area,体表面积
e-r:exposure-response,暴露-效应
pbpk:physiologically based pharmacokinetic,基于生理的药动学模型
pd:pharmacodynamics,药效动力学
pk:pharmacokinetics,药代动力学
poppk:population pharmacokinetics,群体药代动力学
-end-



转载声明:未经本网或本网权利人授权,不得转载、摘编或利用其他方式使用上述作品。已经本网或本网权利人授权使用作品的,应在授权范围内使用,并注明“来源:新领先医药科技”。


 010-61006450
010-61006450 联系地址:
联系地址: 技术市场部邮箱:
技术市场部邮箱: 北京新领先
北京新领先 新领先药讯
新领先药讯 010-61006450
010-61006450
